溶出度(Dissolution rate)也稱溶出速率,是指在規定的溶劑和條件下,藥物從片劑、膠囊劑、顆粒劑等固體制劑中溶出的速度和程度。測定固體制劑溶出度的過程稱為溶出度試驗(Dissolution test),它是一種模擬口服固體制劑在胃腸道中的崩解和溶出的體外試驗方法。藥物溶出度檢查是評價制劑品質和工藝水平的一種有效手段,可以在一定程度上反映主藥的晶型、粒度、處方組成、輔料品種和性質、生產工藝等的差異;在產品發生某些變更后(如處方、生產工藝、生產場所變更和生產工藝放大),確認藥品質量和療效的一致性;也是評價制劑活性成分生物利用度和制劑均勻度 的一種有效標準,能有效區分同一種藥物生物利用度的差異,因此是藥品質量控制必檢項目之一。
一般認為,難溶性 (一般指在水中微溶或不溶) 藥物,因制劑處方與生產工藝造成臨床療效不穩定的藥物以及治療量與中毒量相接近的藥物(包括易溶性藥物),其口服固體制劑質量標準中必須設定溶出度檢查項。另外固體制劑的處方篩選及生產工藝流程制訂過程中,也需對所開發劑型的溶出度做全面考察。一個可行的溶出度試驗法應是在不同時間、地點對同一制劑的溶出度測定或不同的操作者之間的測定都必須達到試驗結果具有良好的重現性。為了達到以上目的,必須對溶出度測定試驗進行 全面充分的研究。 生物藥劑學(BCS)分類(美GFDA ):
第1類:高溶解度一高滲透性 第2類:低溶解度一高滲透性 第3類:高溶解度一低滲透性 第4類:低溶解度一低滲透性
高溶解度:單個制劑能在250mL,pH值1.0~8.0介質中溶解——相當于中G藥典的“微溶”
高滲透性:**生物利用度≥90%
上述分類可以作為設定體外溶出度質量標準的依據,也可用于預測能否建立良好的體內-體外相關性的依據。BSC提示,對于高溶解度、高滲透性(1類)藥物及某些情況下的高溶解度、低滲透性(3類)藥物,其溶出度在0.1NHCL中15min時為85%即可保證藥物的生物利用度不受溶出的限制,即制劑的行為與溶液相似。
溶出度研究試驗主要包括以下內容:(1)溶出介質的選擇,(2) 溶出介質體積的選擇,(3)溶出方法(轉籃法與槳法)的選擇,(4)轉速的選擇,(5)溶出度測定方法的驗證,(6) 溶出度均一性試驗(批內),(7)重現性試驗(批間)等。
近來在新藥審評中發現,部分研究單位在進行溶出度研究時存在一些問題,主要表現在溶出度研究資料過于簡單或溶出度研究內容不夠全面。現予以具體分析,希望能對溶出度研究有一定的幫助。
1、溶出介質的選擇:
介質種類:
水——**常用,良好脫氣水的pH值為5.0~7.0, 適用于溶解度對pH值不敏感的藥物
鹽酸溶液——模擬酸性胃液的介質, 適用于酸中穩定的藥物,濃度一般為0.1~0.01mol/L,pH值一般為1.0~2.2
磷酸鹽緩沖液(PBS)——模擬中等酸性**弱堿性胃液或腸液的介質, 濃度一般為0.05mol/L,pH值一般為4.5~7.6
醋酸鹽緩沖液——模擬中等酸性胃液的介質, 濃度一般為0.05mol/L,pH值一般為3.4~6.0, 醋酸易揮發,導致pH值變化,溶出速率變化、測定時間長的藥物不宜采用醋酸鹽緩沖液作介質 普通制劑
(1)酸性藥物制劑 pH值分別為1.0或1.2、5.5~6.5、6.8~7.5和水; (2)中性或堿性藥物/包衣制劑 pH值分別為1.0或1.2、3.0~5.0、6.8和水; (3)難溶性藥物制劑 pH值分別為1.0或1.2、4.0~4.5、6.8和水; (4)腸溶制劑 pH值分別為1.0或1.2、6.0、6.8和水; 調釋制劑
pH值分別為1.0或1.2、3.0~5.0、6.8~7.5和水
查詢藥物的PKa值,PKa<3.0,為酸性物質;PKa≥3.0為堿性/中性藥物 介質的pH值:
原則:根據體內吸收部位的pH值環境藥物溶解度對pH值的敏感性(pH-溶解度曲線測定:取過量的原料藥,置具塞試管中,分別加PH1.0-8.0的溶出介質適量,使之成飽和溶液,過濾,取續濾液經HPLC法測定準確溶度,計算溶解度并繪制pH-溶解度曲線;觀察曲線與PH的變化情況)
藥物的穩定性來選擇介質的pH值(確保主成分在溶出介質中的穩定性) 2. 溶出體積的選擇
籃法和槳法的溶出體積應在500~1000ml,目前多采用大杯法900-1000ml。除有充足理由。不建議采用該范圍以外的體積。該體積范圍的設定是為滿足藥物的漏槽條件 —— 溶出介質體積應大于藥物全部溶解所需體積的三倍,以保證藥物溶出不受其溶解性影響而設。而對于小杯法是相對小規格固體制劑,當采用紫外法檢測無法滿足測定要求時,從第二法衍生改良而得,截**目前僅我G收載。但該法對于某些活性成分,由于無法滿足溶出度試驗漏槽條件的要求,故建議在新產品溶出度研究與擬定上盡量勿采用該法,解決的辦法:
①加大進樣量濃度**100~200μl。
②調整流動相使主成分保留時間縮短,色譜峰成“尖峰”狀,積分將更加準確、精密度自然提高。
③不選**大吸收波長,選擇末端強吸收處波長作為檢測波長,以加大響應值。 3.溶出方法裝置和轉速的選擇
籃法和槳法是目前通用性**強、耐用性**高、儀器**為普及的法定溶出方法,因此,建議**該兩法,槳法—**,只要藥物不上浮就應選槳法,推薦50轉/分,一般選用50~75rpm,不推薦采用100轉甚**更高的轉速,因為這將嚴重降低對于不同來源同一制劑的區分力,且大大降低體內外相關性,除非在已驗證該制劑體內具有良好生物利用度的前提下,并應提供充足的理由與驗證資料;轉籃法推薦100轉/分,一般選用50~100rpm;槳法通常將轉籃法/100轉強度看作與槳板法/50轉相當、且將該強度看作與中老年人體的胃腸道蠕動強度等同;當采用槳法/75rpm試驗條件難以準確評價質量時,可改為籃法/120rpm。但是在某溶出介質中,當結束時間點溶出量仍達不到90%,緩/控85%時,可放寬溶出參數,如現增加轉速75轉/分,未果的話可以加入表面活性劑。 4. 測定時間點和結束時間點的設定 測定時間點:
普通和腸溶制劑可分別為5、10、15、20、30、45、60、90120min,此后每隔1小時測定
緩/控制劑:15、30、45、60、90分鐘和2、3、4、5、6、8、10、12、24小時 結束時間點:
在酸性介質(ph1.0-3.0)中**長為2h,其他PH介質中,普通和腸溶為6h,緩/控為24h,擔當連續兩點達90%(緩/控85%),且差值小于5%時,可提前結束。 5. 取樣時間點和限度的擬定
高溶解度且快速溶解產品:單個時間點,大多數情況。對于普通制劑,建議以**次出現溶出量均在85%(或90%)以上的兩時間點,且該兩點溶出量差值在5%以內,取前一時間點作為質量標準中的取樣時間點,并將該點的溶出量減去15%作為溶出限度。取樣時間一般為5的倍數,3以上可向上約靠。
低溶解度且溶出緩慢的制劑或對溶出度有特殊要求的制劑:兩個時間點 緩釋制劑:藥物作用8小時的一般**少設3個時間點
作用12~16小時的**少設4個時間點 作用24小時或以上的**少設5個時間點
6. 含量測定方法的選擇與驗證
原則:專屬好、重復性好、方便快捷、易于自動化
盡可能與含量測定方法相同
紫外分光光度法——**
無紫外吸收的,可衍生化后比色
溶出度測定普遍采用方便快捷的UV法,但一定要注意輔料的干擾。特別是在短波長處,更應注意輔料的末端吸收等干擾因素的存在。一般認為輔料的干擾應在2%以內,否則UV法不適用。
采用UV法測定前,一定要進行紫外掃描,以考察輔料是否存在干擾。方法為:分別取對照品溶液和供試品溶液(濃度**好與對照品溶液一致或略低),進行紫外掃描。著重觀察供試品溶液圖譜情況:是否存在基線被抬升的現象,以及與對照品溶液圖譜重合的情況。如輔料無干擾,則供試品溶液圖譜應與對照品溶液圖譜基本重合或整體略低于對照品圖譜;如輔料有干擾,就會出現基線或末端
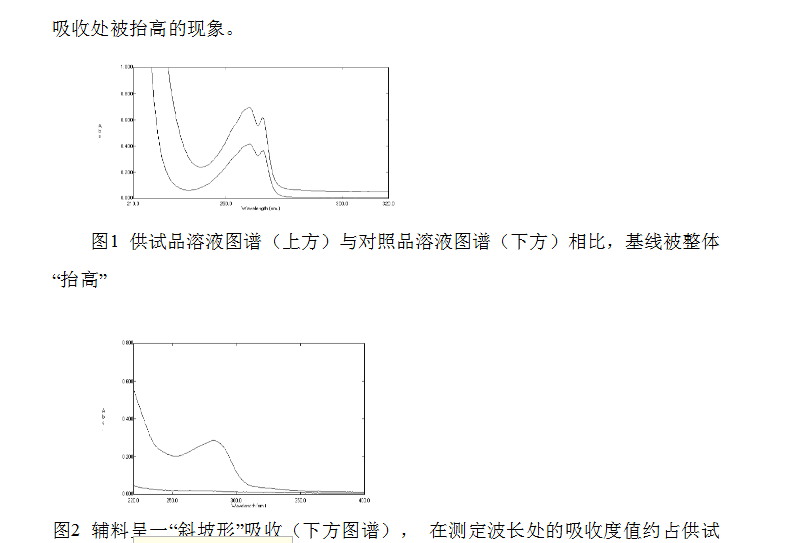
品溶液(上方圖譜)的5%左右
另一方法可采用HPLC法,將其測定結果與UV法進行比較,以判定輔料是否有干擾。
當輔料干擾紫外法測定時,通常可采用以下方法:
雙波長吸收度差值法: 導數光譜法:
HPLC法:在HPLC色譜柱上可輕松將輔料與主成分分離,該法測定**為準確、可靠,只是工作量略顯增加。
驗證:
溶出度檢測方法按照既定的方法學認證各項要求驗證,尚需注意以下各事項:
(1) 對于某些難溶性藥物,如難以采用溶出介質直接溶解對照品,可先加少量有
機溶劑(如甲醇或乙醇)溶解后,再加溶出介質稀釋的辦法,建議**終溶液中有機相比例不超過5%。如超出,應予以相應驗證。
(2) 線性范圍應考慮到有可能出現的低濃度點情況,如對于溶出曲線或調釋制劑的測定等。并為減小外標一點法的測定誤差,建議將對照品溶液配制為約50%釋放量濃度。
(3) 應注意考察活性成分在37℃溶出介質中的穩定性。若不佳,應在質量標準中
標注“取出后立即測定的”字樣;不建議在溶出介質中加入抗氧劑、穩定劑等物質。
(4) 根據實際情況,檢測波長可選取非**大吸收波長。
(5) 當測定法為紫外法時,由于易受輔料或膠囊殼的影響,建議分別取對照品溶
液與供試品溶液進行紫外掃描后根據所得圖譜予以明了,亦可采用高效液相法進行佐證。如干擾存 在,可考慮采用雙波長相減法或空白膠囊殼扣除法等方法予以消除,不建議采用紫外導數光譜法;如干擾排除較為困難,建議采用高效液相色譜法測定。
(6) 空白膠囊殼干擾在2%以內可忽略不計;若大于2%則應校正,并在該品種項
下予以注明;大于25%時,則被視為溶出試驗無效,應重新選擇測定方法。關于其測定法,建議取6粒樣品,徹底清除其中內容物,同法試驗和過濾后,分別量取相同體積混勻測定。測定時、以相應溶劑為空白。
(7) 不應出現溶出度測定結果均值高于含量測定結果3%以上的情況。如出現,應
重新考察溶出度測定方法的適用性。
(8) 對于小規格制劑,不建議采用大于1cm的吸收池進行紫外法測定,而建議采用HPLC法(并可酌情加大進樣量以提高測定精密度)。但對于具有較強紫外吸收的活性藥物,為提高工作效率,可在溶出曲線大批量樣品測定時,采用0.1cm~1.0cm短吸收池,但必須經過驗證。
(9) 對一些小規格制劑,樣品過濾時會發生吸附,判定吸附與否的方法可采用:
①取溶出液,分別過濾不同體積的初濾液后測定,觀察響應值的變化情況, ②取樣后一份不過濾,直接采用高速離心,取上清液測定。并與采用過濾法所得的續濾液比較,考察兩者間測定數據的差異。判定自動取樣
溶出儀的固有濾膜是否存在吸附時,一般采用該法。
③取對照品溶液,經濾膜過濾后,與原溶液進行比較,觀察測定前后數據的變化情況。
如濾膜對藥物有吸附,可采用將濾膜在沸水中煮沸1h,或加大初濾液體積等辦法予以解決。但建議采用直接高速離心的方法。 7. 溶出曲線比較
產品上市發生較小的變更時,采用單點溶出度試驗可能就足以確認其是否未改變產品的質量和性能。發生較大變更時,則推薦對變更前后產品在相同的溶出條件下進行溶出曲線比較。在整體溶出曲線相似以及每一采樣時間點溶出度相似時,可認為二者溶出行為相似。
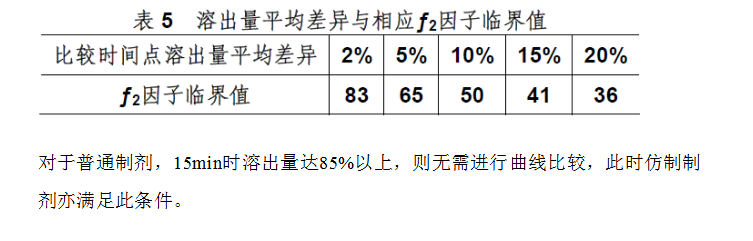
由于多pH值溶出曲線的繪制已成為剖析和表達固體制劑內在品質的重要手段,故對溶出曲線比較的科學評價愈發重要。截**目前,報道有多種比較方法。
但自美G和日本等G的官方機構認定采用模型非依賴方法之一的“相似因子比較法”之后,現基本上被統一采納。該法特點是對溶出曲線進行整體評價,通過計算相似因子(ƒ2
)比較溶出行為的相似性。
通常,當ƒ2
數值介于50-100認為兩條溶出曲線是相似的,該數值限定是基于
兩條比較曲線上任一比較時間點溶出量平均差異限度不大于10%的考慮。表5提供了溶出量平均差異與相應ƒ2
因子臨界值的關系。